
As described in last months’ articles, genomic methods are increasingly and effectively used to support diagnostic, preventive, and therapeutic strategies and enhance the development of personalized medicinal approaches.
Please find our previous articles here: Genomic Corner
Evolution of genomics
Genomics is the study of the structure, function, and evolution of genomes that has seen a dramatic development following the realization of the Human Genome Project and the continuous progress of DNA sequencing technologies.
The Human Genome Project aimed to identify the complete sequence of the human genome, took over a decade to complete (1990–2003), involving more than 20 institutes worldwide and costing about 3 billion dollars [1,2]. The completion of the project at the beginning of this century revolutionized biotechnology. After the first draft of the human genome sequence was unveiled, continuous information was generated, shedding light on the enclosed genes, the biological functions of their products, and their involvement in diseases.
Technological breakthroughs over the past decades have resulted in the drastic increase of DNA sequencing processes’ content and automation level while reducing the cost per base (Figure 1). The first standard method for DNA sequencing, i.e., Sanger sequencing, was developed in the 1970s, allowing the analysis of few DNA fragments at a time (length < 1000 bp) [3]. The method was very accurate, still a golden standard, but too expensive and laborious if applied to large or multiple targets for genomic studies. Subsequently, the introduction of next-generation sequencing (NGS)-based methods, which have progressively allowed the parallel sequencing of multiple genes, the whole protein-coding region (whole-exome sequencing, WES) or genomes (whole-genome sequencing, WGS) [4].
In this century, NGS-based methods have dramatically advanced the identification of the genetic causes of Mendelian phenotypes, providing support for diagnostic, preventive, and therapeutic strategies, and supporting the development of personalized medicinal approaches is expected in upcoming years (1, 2). Here, we want to highlight some of the WES-based approach’s achievements in specific diagnostic fields.
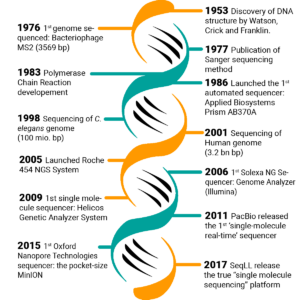
Figure 1. Timeline of the most remarkable milestones in the progress of DNA sequencing [5].
Next-generation sequencing (NGS) defines massively parallel sequencing technology that in the last decades has revolutionized genomic research. NGS provides high-throughput and scalable methods supporting a wide set of applications in research and diagnostic.
Gene panels are tests targeted to the analysis of a set of genes or genetic regions or variants that have known or suspected associations with the studied disease or phenotype.
Whole-genome sequencing (WGS) is a sequencing technique for the analysis of the entire genome.Whole-exome sequencing (WES) is a sequencing technique for analyzing all the protein-coding regions in the genome (i.e., exome).
Nucleotides (WGS) are the monomers of DNA strands.hg19 and hg38 and hg38 are different assemblies of the human reference sequence (respectively, Genome Reference Consortium Human Build 37 and Genome Reference Consortium Human Build 38).
On the side of the progress, the “genomic revolution” has brought high demand in terms of processing, analysis, and interpretation of the massive amount of continuously generated data [6]. The increasing size and complexity of the derived information progressively lessen the efficiency of manual processes, requiring sophisticated applications of computer technology to process the generated data in a reasonable time meaningfully.
Bioinformatics in genomics
The computer methods and programs used to understand and use of biological and biomedical data are referred to as Bioinformatics. This covers acquiring, storing, analyzing, and interpreting biological data (e.g., DNA sequences). Bioinformatics is crucial in driving genomics knowledge and can be seen as a multidisciplinary field featuring knowledge from biology, computer science, mathematics, statistics, and medicine [7,8,9,10].
All NGS processes, regardless of the application and platform used, can be outlined in library preparation and sequencing, followed by the analysis of the generated data. NGS data analysis typically includes three main processes: base calling (primary analysis), read mapping and variant identification (secondary analysis), and variant annotation (tertiary analysis) [11].
We present an overview of the different steps involving bioinformatic analysis of NGS data, referring to generic genomic sequencing in humans, being mindful that different sequencing and analytical applications might require specific workflows and settings.
Primary data analysis
The analysis starts with calling the base pairs and their quality generated by the sequencing process, resulting in DNA sequences (i.e., strings of A, C, G, and T nucleotides) enclosed in the sequencing output file. Since the quality of the raw data is crucial for the overall outcome of the analysis, each base call is listed with the quality score of the call to be evaluated and used in the next analysis steps.
As NGS methods allow multiple samples to be sequenced, the demultiplexing process – following the sequencing – allows attributing sequences to each sample using the barcode information. The primary analysis is performed by the sequencing platform with machine-specific methods and software.
Secondary data analysis
The reads generated by the sequencing are mapped to a reference genome to identify in the DNA sequence, so-called variants. Beforehand, the raw data’s quality control (QC) is performed to ensure suitable quality for the downstream processes [6].
Quality Control reads filtering and trimming
Low-quality reads will probably align poorly due to potential mistakes in base calling or map to multiple or wrong places in the genome. Therefore, discarding it might improve the overall results.
The filtering can be done based on quality scores expressed as logarithmic error probability (Phred score). For instance, Q10 means that a base has 1 out of 10 probability of not being correct (i.e., an accuracy of 90.0%), and Q30 1 out of 1000 probability of not being correct (i.e., 99.9% accuracy) [12]. Trimming is performed at the ends of each read to remove residual adapters’ sequences that may interfere with the downstream analysis processes.
Filtering and trimming, although reducing the overall number and length of the reads, increase the quality and the reliability of the results [5].
Reads mapping
Once a suitable level of quality is reached, the sequenced fragments can be aligned against a reference genome (reads alignment) or used to assemble a genome from scratch without the help of external data (de novo assembly). For most NGS applications in human genomics, e.g., clinical genetics, the most common choice is the alignment against the human reference genome (hg19 or hg38).
The alignment is a prime example among the problems managed by bioinformatics. As the sequencing reads produced by most of the NGS platforms are short, millions of DNA fragments need to be matched and mapped to a reference, like a puzzle that would take forever for a single person to execute manually (Figure 2).
A crucial point in this process is setting a line between what could be considered an actual variation and what can be considered a misalignment. Different factors could complicate the process, as sequencing errors and actual discrepancies between the sequenced results and the reference genome could determine misalignment issues. Besides, the computational challenge is increased by the complexity of different regions of the genome, as it contains many repetitive elements and very homologous areas. In certain conditions, it is tough to determine to which repeat copy the read belongs or to which position the reads correctly align [5].
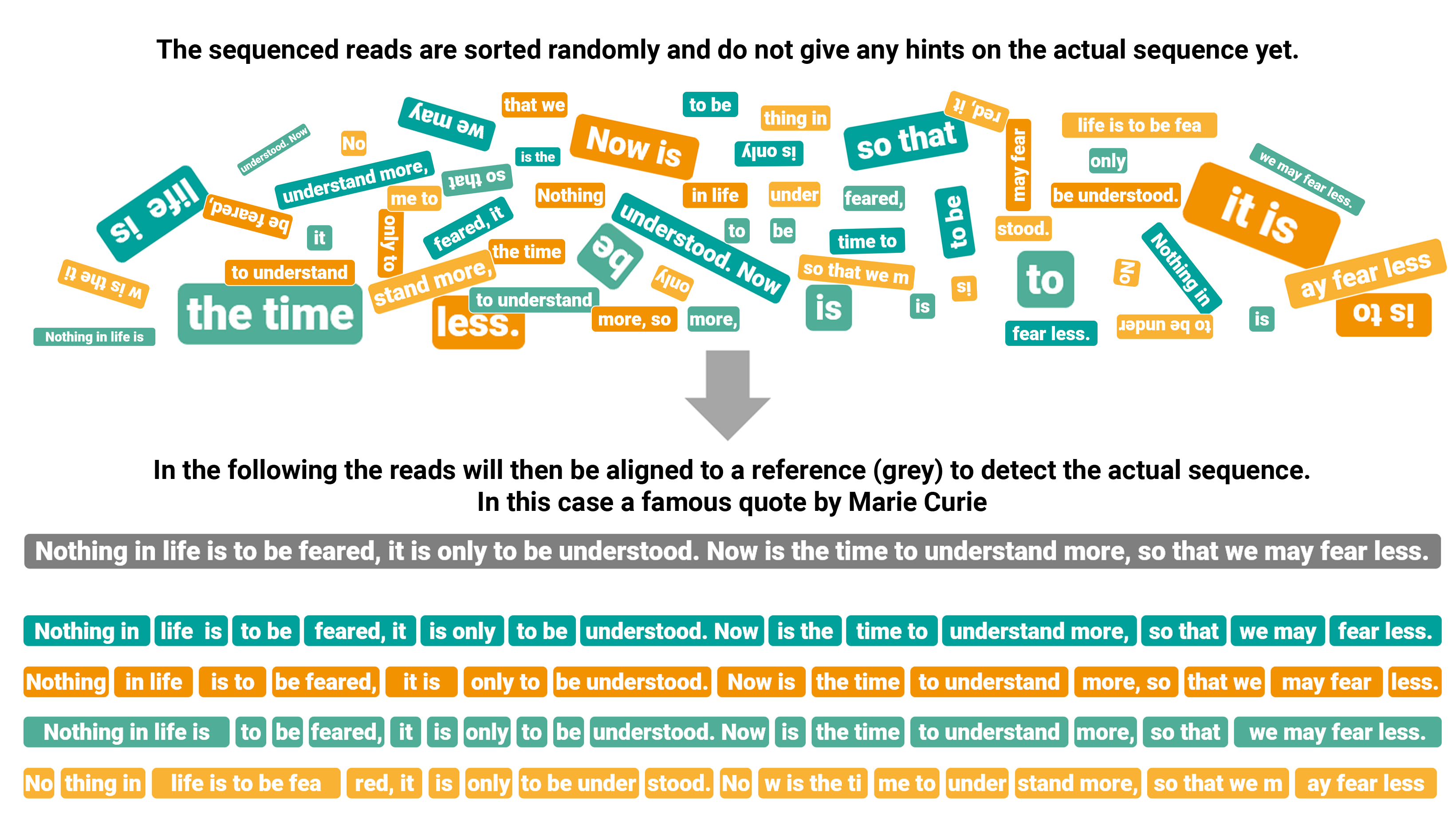
Figure 2. Graphical transposition of reads alignment.
Variant identification
Variant calling is the process of identifying variants from the sequenced data. This process is aimed to recognize where the aligned reads differ from the reference genome and write the identified variants to the final result file.
Different types of variants can be identified according to the selected application.
In general, we can view genetic variation from the nucleotide-level to the chromosome-level perspective. The changes at the nucleotide level are defined as single-nucleotide variants (SNVs). Small insertions or deletions (indels) represent gains or losses of genetic sequences usually smaller than 50bp, while structural variations (SVs) include larger differences (even ≥1kb). Copy-number variants (CNVs) are duplications or deletions of DNA segments present in a number of copies different from the reference. Other macroscopic forms of structural variations can affect the number and/or the genetic content of one or more chromosomes. Based on the number, SNVs represent the largest category of variations, taking place on average each 8bp within the exonic regions according to updated estimates [13,14]. From a technical perspective, the detection of some forms of variations remains problematic, including small CNVs (<500bp), the insertion of sequences different from the reference, subtle variations in the number of copies, highly-represented dispersed repeats (e.g., Alu elements, LINEs) and CNVs located on the Y chromosome or heterochromatic regions [15,16].
Tertiary data analysis
After the genomic variants are detected, the last analytical effort is “making sense” of the found variants and supporting the data’s interpretation. In the context of clinical human genetics, this means finding the biological significance of the identified variants in order to connect them with the observed phenotype.
The tertiary analysis begins with assigning a series of information to the variants founded with the previous analysis steps to help predict their functional impact (i.e., variant annotation) [17].
Many different layers of information and resources can be used, commonly starting from databases listing variants that have been previously reported and described (e.g., clinical significance, population-level frequency, prediction of the exerted consequences, etc.).sed on the annotation, variants can be subsequently filtered and prioritized [5].
By Luca Trotta on September 29, 2021
[1] Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017;18(1):14. Published 2017 Feb 14. doi:10.1186/s12863-017-0479-5[2] Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The Sequence of the Human Genome. Science 2001 American Association for the Advancement of Science;291(5507):1304-1351.
[3] Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 1975 May 25;94(3):441-448.
[4] Jamuar SS, Tan EC. Clinical application of next-generation sequencing for Mendelian diseases. Hum Genomics 2015 Jun 16;9:10-015-0031-5.
[5] Pereira R, Oliveira J, Sousa M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J Clin Med. 2020 Jan 3;9(1):132. doi: 10.3390/jcm9010132. PMID: 31947757; PMCID: PMC7019349.
[6] Morganti S, Tarantino P, Ferraro E, D’Amico P, Viale G, Trapani D, Duso BA, Curigliano G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit Rev Oncol Hematol. 2019 Jan;133:171-182. doi: 10.1016/j.critrevonc.2018.11.008. Epub 2018 Nov 26. PMID: 30661654.
[7] Luscombe NM, Greenbaum D, Gerstein M. What is bioinformatics? A proposed definition and overview of the field. Methods Inf Med. 2001;40(4):346-58. PMID: 11552348.
[8] https://www.britannica.com/science/bioinformatics
[9] https://www.genome.gov/genetics-glossary/Bioinformatics
[10] https://www.merriam-webster.com/dictionary/bioinformatics
[11] Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit Rev Oncol Hematol. 2019 Jan;133:171-182. doi: 10.1016/j.critrevonc.2018.11.008. Epub 2018 Nov 26. PMID: 30661654.
[12] Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185.
[13] 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature 2015 Oct 1;526(7571):68-74.
[14] Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016 Aug 18;536(7616):285-291.
[15] Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008 Apr 17;452(7189):872-876.
[16] Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature 2010 Apr 1;464(7289):704-712.
[17] Chakravorty S, Hegde M. Gene and Variant Annotation for Mendelian Disorders in the Era of Advanced Sequencing Technologies. Annu Rev Genomics Hum Genet. 2017 Aug 31;18:229-256. doi: 10.1146/annurev-genom-083115-022545. Epub 2017 Apr 17. PMID: 28415856.
Picture: gagnonm1993 / pixabay

