
Next-generation sequencing (NGS) has revolutionized patient care in the last decades, and we saw here how effective it has proved for the prevention, and therapeutics of rare disorders. Similarly, in the field of cancer care, NGS-based methods have contributed to delivering unprecedented advances in understanding the biology of diseases and led to influential clinical implications.
In anticipation of World Cancer Day on February, 4th (tomorrow), we want to provide you with information about cancer and how it relates to genomics. We at GenomSys hope – with this article – to contribute to raising public awareness on the prevention, research and treatment of cancer.
What is cancer?
What we commonly define as cancer can be intended as a diverse spectrum of human diseases. More than 100 different types of cancer are known, mostly named from the tissue where they arise from.
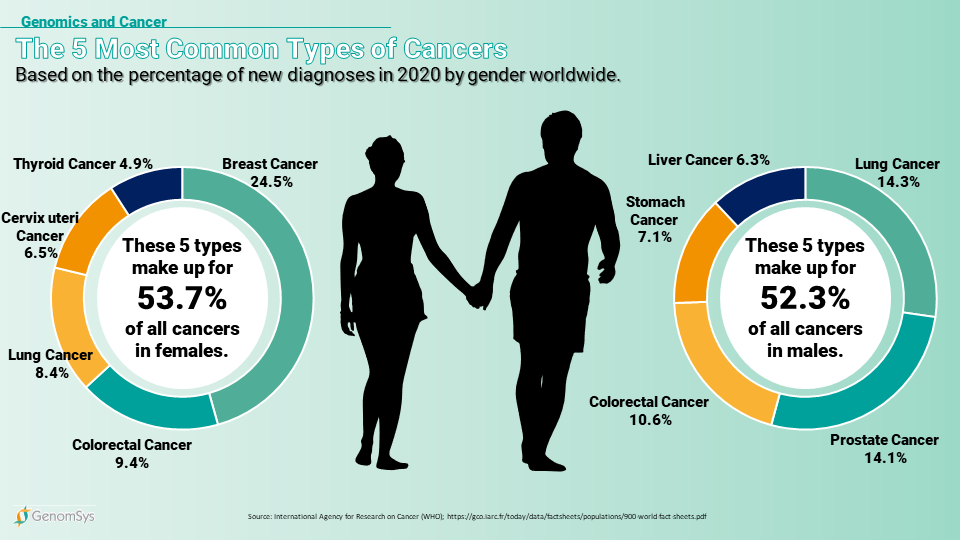 Graphic 1 – The most common types of Cancers in 2020 for each gender worldwide (18).
Graphic 1 – The most common types of Cancers in 2020 for each gender worldwide (18).
Among the most common cancers and their root tissues in adults:
- carcinomas (derived from epithelial cells lining body cavities and glands),
- sarcomas (from the connective tissues),
- melanomas (from pigmented cells of the skin),
- retinoblastomas (dividing cells in the ocular retina),
- neuroblastomas (from immature peripheral neurons),
- glioblastomas (from glia, i.e., neural support cells) and
- lymphomas and leukemias (i.e., “liquid tumors”, arising in the tissues that originate lymphoid and blood cells).
When cancer arises, the symptoms and the possible treatment depend on the type of disease and advancement stage. Treatment strategies can include surgery, radiation, chemotherapy, immunotherapy or other types of biologic therapy, hormone therapy, or stem cell transplantation (1,2).
The Cancer Research charity in the UK has a broad database with definition of each cancer type. Find out more here.
How many people are worldwide affected by Cancer today and what is the outlook?
In 2020 around 19.3 million new cancer cases were registered. Today Cancer is the second-leading cause of death worldwide and the costs of cancer are estimated at US$1.16 trillion. The International Agency for Research on Cancer (IARC) estimates that by 2040 new cancer cases are expected to grow to 30.2 million cases solely due to the growth and aging of the population(18, 19).
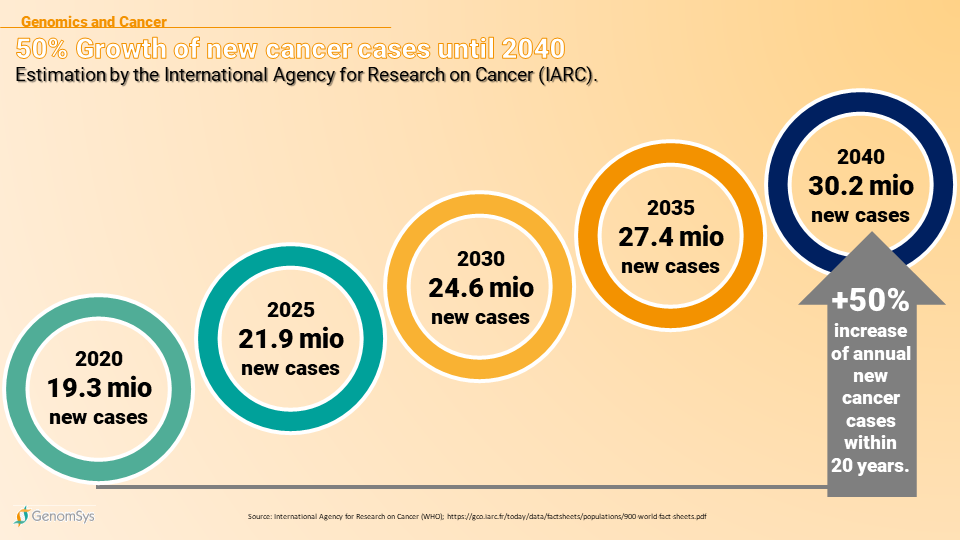
Graphic 2 – Estimated development of cancer in the next 20 years worldwide (18).
The International Agency for Research on Cancer, a part of the World Health Organization (WHO) developed an interactive tool to learn more about cancer statistics. Find out more about the tool here.
What is the cause of cancers?
The body’s building blocks are cells, over 200 different types organized into tissues and organs.
New cells generate as needed by the body to replace the old ones that die or stop working. When some of these processes mess up, new cells could grow albeit don’t need, and old or malfunctioning cells could escape death, and the resulting extra cells can amass into tumors. Conversely, from benign tumors, cells from malignant ones can breach nearby tissues or breakaway, spreading throughout the body (metastasis) (2). Different factors, including environmental, can contribute to cancer; however, the fundamental concept underlining this complexity is the presence of alterations in the genes. In the last two decades, a revolution in genomic methods has allowed us to profoundly deeply investigate the meaning of human genetic variation and its possible role in triggering the onset and growth of tumors (1).
The Cancer Treatment Centers of America – CTCA made a very well explained video on what cancer is and how it is caused. Check out the video here.
How common are cancers caused by variations of genes?
Cancers due to mutations in our DNA are less common than cancers caused by environmental factors like ageing or behavior. Experts estimate that up to 10% of detected cancers are linked to an inherited faulty variation within specific genes. Depending on the affected gene it can define the type of cancer as well as the risk level for developing this disease (19).
How can genomics help in cancer care?
DNA alteration occurring in a body’s reproductive cell (egg or sperm) that becomes incorporated into the DNA of every cell in the offspring’s body are defined germline. (3). Somatic variation represents DNA alterations occurring after conception, and therefore not present within the germline and not passed on to offsprings (4).
Deciphering germline and somatic variation can provide meaningful information for various purposes supporting diagnostics, prognosis, treatment selection, and drug response (5, 6,7).
Germline analysis can be used to tackle hereditary cancer syndromes, while the analysis of the somatic genome (from tumor specimens or liquid biopsies) can drive molecularly guided therapy in oncology (6). Additionally, somatic and germline samples can be compared to increase the sensitivity in low-quality tumor samples and unambiguously depict germline mutations (7).
Examples of NGS application in oncology:
The main focus of genetic testing in oncology is on inherited cancer syndromes, cancer somatic mutation analysis, and pharmacogenomics.
- diagnostic (e.g., for hereditary breast and ovarian cancer) (8),
- hereditary risk assessment (e.g., Lynch syndrome) (9),
- prognosis (e.g., KRAS mutations in lung adenocarcinoma) (10),
- treatment selection (e.g., biomarkers for immunotherapy responsiveness such as tumor mutational burden and microsatellite instability) (11),
- therapeutic selection for clinically actionable alterations (e.g., BRAF V600E in colorectal cancer) (12).
What are hereditary cancer syndromes?
We talk about hereditary cancer syndromes when a higher-than-normal risk of a certain type of cancer runs in the family. The risk originates from mutations that can be passed from parents to children [13]. Most known examples of hereditary cancer syndromes are hereditary breast and ovarian cancer syndrome, Lynch syndrome, Li-Fraumeni syndrome (14).
Certain patterns may be depicted within families, helping to address hereditary cancer syndrome (e.g., the presence of the same type of cancer in multiple family members even from different generations, development at unusual or early age, the occurrence of multiple types of cancer in the same person) [15].
The genetic predisposition of many cancer syndromes has been unraveled, and diagnostic screening programs currently target the discovered and responsible genes. The analysis of mutation in genes associated with specific familial cancer can augment the chance of survival and help define prognosis and personalized treatment or preventive strategies [6, 14].
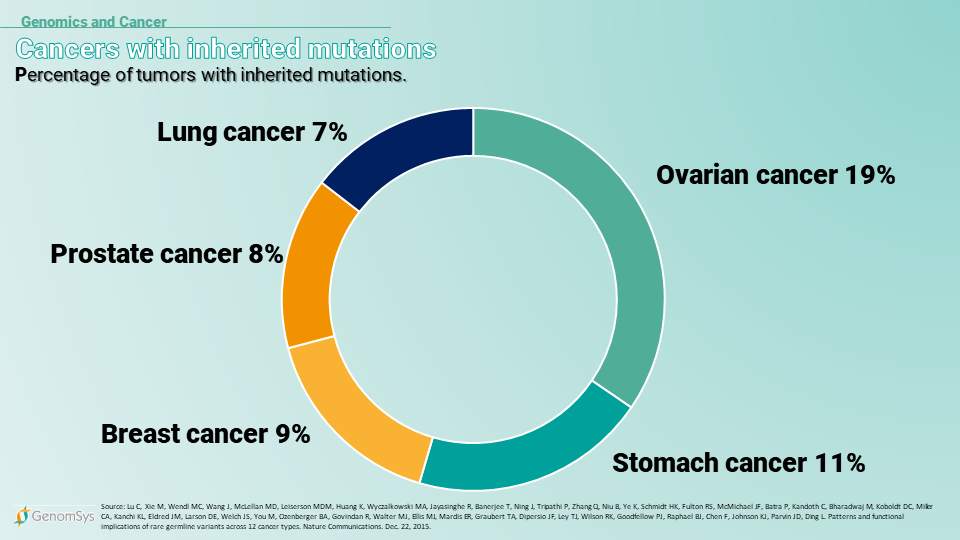
Graphic 3 – Percentage of cancer cases where an inherited mutation is a factor (20).
Which types of genetic testing are used when inherited cancer syndromes are suspected?
NGS panels focused on established cancer genes frequently represent a cost-efficient solution due to the relatively lower demand in terms of data handling, storage, and analysis. Additionally, restricting the sequencing to the high risk-genes only (targeted approach) decreases the chance of finding variants of unknown significance (VUS), which are difficult to interpret and use in clinical management.
Currently, the reduction of analysis costs has made the use of WES and WGS more applicable and probably a preeminent approach in the near future.
Lastly, besides the screening of the genomic DNA, novel approaches such as RNA-seq can augment the power to identify genetic causes of cancer [6].
Why are cancer somatic mutations studied?
WES/WGS studies have primarily contributed to unraveling the mutational landscape in cancer, and now NGS methods are widely applied to scan somatic alteration for diagnostic and therapeutic purposes.
Targeted screening of somatic variants, including single nucleotide variants, indels, copy number variations (CNVs), and large genomic rearrangements (structural variants), can be used for clinical applications, improving patient classification, prognosis prediction, targeted treatments, drug resistance, and pharmacogenetics. Different approaches can be used for the identification of the genetic aberrations underlying tumor initiation, development, and metastasis, driver genes, driver and passenger mutations, either by comparing the somatic mutation pattern in cancer biopsies with that observed in germline DNA from the same patients (or from a healthy tissue) or by comparing with reference DNA (6).
What is Pharmacogenomics?
Pharmacogenomics (PGx) studies the role of person’s genes on their response to medications (16). Many drugs don’t work the same way in different persons., The differences in terms of effectiveness and toxicity are attributed to a variety of factors such as environmental, diet, lifestyle, disease conditions and co-medication, and individual genetic makeup. Variation in genes encoding enzymes or proteins involved in drug pathways can influence the individual drug response or susceptibility to adverse drug reactions (ADRs) (6, 17).
Several PGx applications focus on anticancer agents due to the variable efficacy of treatments and the toxicity of chemotherapeutics. In certain types of cancer, the tumor genome can be screened for biomarkers with predictive or prognostic meaning to be used for targeted treatments.
The NGS-based approaches for PGx can involve the analysis of the entire exome (WES), genome (WGS), or targeted panels. WGS offers the advantage of encompassing also the non-coding regions, while diagnostic PGx applications mainly focus on one or a few genes related to the use of one specific drug (or several ones metabolized via the same pathway) (6).
By Luca Trotta on February 03, 2022
[1] Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017;18(1):14. Published 2017 Feb 14. doi:10.1186/s12863-017-0479-5[2] Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The Sequence of the Human Genome. Science 2001 American Association for the Advancement of Science;291(5507):1304-1351.
[3] Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 1975 May 25;94(3):441-448.
[4] Jamuar SS, Tan EC. Clinical application of next-generation sequencing for Mendelian diseases. Hum Genomics 2015 Jun 16;9:10-015-0031-5.
[5] Pereira R, Oliveira J, Sousa M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J Clin Med. 2020 Jan 3;9(1):132. doi: 10.3390/jcm9010132. PMID: 31947757; PMCID: PMC7019349.
[6] Morganti S, Tarantino P, Ferraro E, D’Amico P, Viale G, Trapani D, Duso BA, Curigliano G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit Rev Oncol Hematol. 2019 Jan;133:171-182. doi: 10.1016/j.critrevonc.2018.11.008. Epub 2018 Nov 26. PMID: 30661654.
[7] Luscombe NM, Greenbaum D, Gerstein M. What is bioinformatics? A proposed definition and overview of the field. Methods Inf Med. 2001;40(4):346-58. PMID: 11552348.
[8] https://www.britannica.com/science/bioinformatics
[9] https://www.genome.gov/genetics-glossary/Bioinformatics
[10] https://www.merriam-webster.com/dictionary/bioinformatics
[11] Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit Rev Oncol Hematol. 2019 Jan;133:171-182. doi: 10.1016/j.critrevonc.2018.11.008. Epub 2018 Nov 26. PMID: 30661654.
[12] Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185.
[13] 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature 2015 Oct 1;526(7571):68-74.
[14] Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016 Aug 18;536(7616):285-291.
[15] Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008 Apr 17;452(7189):872-876.
[16] Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature 2010 Apr 1;464(7289):704-712.
[17] Chakravorty S, Hegde M. Gene and Variant Annotation for Mendelian Disorders in the Era of Advanced Sequencing Technologies. Annu Rev Genomics Hum Genet. 2017 Aug 31;18:229-256. doi: 10.1146/annurev-genom-083115-022545. Epub 2017 Apr 17. PMID: 28415856.
[18] International Agency for Research on Cancer (WHO); https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf
[19] Cancer Research UK, https://www.cancerresearchuk.org/about-cancer/causes-of-cancer/inherited-cancer-genes-and-increased-cancer-risk/family-history-and-inherited-cancer-genes
[20] Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MDM, Huang K, Wyczalkowski MA, Jayasinghe R, Banerjee T, Ning J, Tripathi P, Zhang Q, Niu B, Ye K, Schmidt HK, Fulton RS, McMichael JF, Batra P, Kandoth C, Bharadwaj M, Koboldt DC, Miller CA, Kanchi KL, Eldred JM, Larson DE, Welch JS, You M, Ozenberger BA, Govindan R, Walter MJ, Ellis MJ, Mardis ER, Graubert TA, Dipersio JF, Ley TJ, Wilson RK, Goodfellow PJ, Raphael BJ, Chen F, Johnson KJ, Parvin JD, Ding L. Patterns and functional implications of rare germline variants across 12 cancer types. Nature Communications. Dec. 22, 2015.
Picture: gagnonm1993 / pixabay

