
In this week’s article from our series “MPEG-G, can I eat it?” we would like to explain how the standard can support more efficient processing of genomic datasets through MPEG-G’s Selective Access benefit.
If you missed our previous articles, you could find them here:
Finding the variant amongst a 3-billion-pieces Haystack takes time
The growing insights in genomics over the past decade have made genomic testing a valuable tool in healthcare. While we have steadily increased our knowledge of these 3 billion building blocks of our DNA, we fully understand still “only” about 2% of this archive of information that is responsible for life on this planet.[1]
Today through Next-Generation Sequencing (NGS) and the extraordinary efforts of the Human Genomics Project, we can sequence our entire DNA within a day. The massive parallel sequencing of multiple genes, the whole protein-coding region (Whole-Exome Sequencing, WES) or genomes (Whole-Genome Sequencing, WGS) with NGS is now the technological cornerstone for the sequencing, transforming biological information stored in our DNA into a digital format.[2]
The increasing adoption of genetic testing is both propelling and challenging the implementation of analysis methods. The plethora of generated data fundamentally helps in the scientific progress for genomics. Newfound disease-causing variants can shed light on the biology of disorders and be of use for diagnostics and therapeutics. On the other hand, it has led to a bottleneck in the analysis. Although the data is now faster available, the analysis is very time-consuming. Existing data formats in genomics are also reaching their limits in analyzing this vast amount of genomic data. The gained speed of sequencing for the entire genomic testing is thus slowed down by the digital data processing, which can delay the delivery of findings and the start of the treatment.
Additionally, a specific issue arose in dealing with this vast amount of genomic data. The risk for incidental findings or secondary findings, being results unrelated to the initial indication for genomic testing, might increase and thus confront geneticists in their daily analysis with the ethical question on how to deal with these findings.[3]
Among other things, the MPEG-G standard aims to help eliminate this bottleneck and provide a basis for any future technological solutions in genomics. The integrated Selective Access feature of the unique ISO standard helps to process genomic data faster. Thus, it allows the geneticist to initiate their analysis of the results earlier and decrease the risks for incidental findings to zero.
Selective Access – What is it? And How does MPEG-G improve it?
Selective Access or Random Access, as it is called in computer science, is the ability to access data selectively instead of sequentially. This feature is vital for accessing specific regions – chromosomes, genes, or even single bases – on the DNA within large sequencing datasets, as they are common with WES or WGS, to analyze the data faster and without waiting for sequential processing before starting.[4]
During the creation of MPEG-G, particularly during part 1, the Selective Access feature was developed to provide a data format that needs significantly less memory and processing time to access compressed required data for each analysis. The core lies within the indexing structure of MPEG-G, which is natively embedded with the data. The indexing step and the sorting step – although sorting in MPEG-G is not strictly required – are an inherent part of the encoding process. Hence they can be performed concurrently with the alignment, for example, without any overhead in terms of processing time: this allows later rapid access to the desired genomic region without the need to use external indexing information to be built before accessing the data. (Edited 17.03.2022)
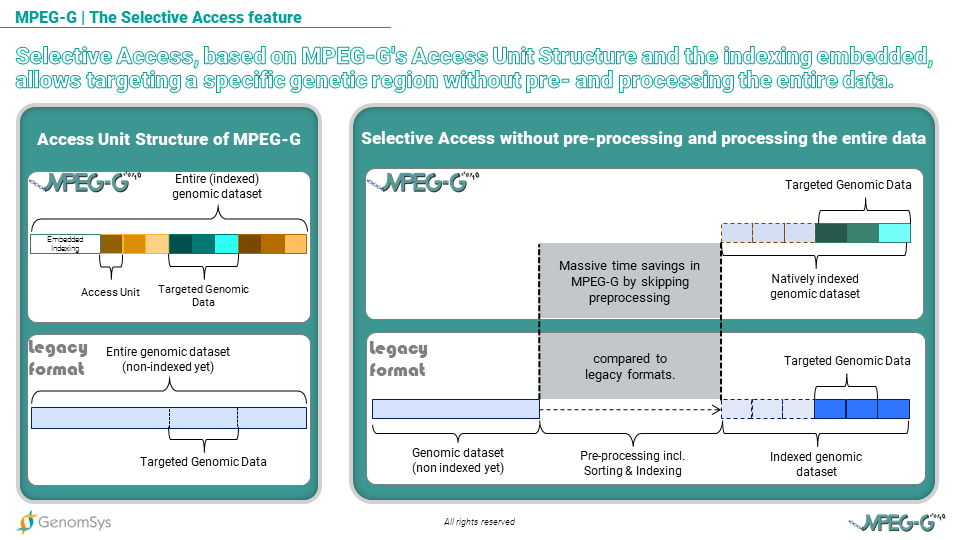
Graphic 1 – Selective Access, based on MPEG-G’s Access Unit Structure and the indexing embedded, allows targeting a specific genetic region faster than legacy formatted genomic data.
“
MPEG-G Selective Access is based on access units structure and embedded indexing. This approach enables focusing on specific genetic targets without accessing and decoding the entire data. It also strongly accelerates data processing by completely removing the need for pre-processing stages and by enabling parallel processing and data flow,” explains Head of Genomic Data Engineering at GenomSys Daniele Renzi.
About Daniele Renzi
Daniele still is and was in the past a significant contributor of MPEG-G, the ISO/IEC standard for genome sequencing data representation. After receiving an engineering master’s degree from the Polytechnic University delle Marche (Italy), he participated in several standardization activities within ISO/IEC in multiple domains such as multimedia systems, video coding, and genomics. In his career, Daniele designed and developed solutions for the digital multimedia market and the genomic market. In GenomSys, he leads the design and development of the MPEG-G Codec Suite.
Also, with MPEG-G genomic data can be transmitted with minimum delay between data generation and consumption thanks to its access unit structure. So each access unit is an independent data chunk and can be as small as needed, making the consumer able to process data as soon as the first access unit is received, thus minimizing the overall data processing time in the genomic pipeline. This “transport feature”, which is similarly used in streaming videos today by companies like Netflix, avoids being stuck with long waiting times between different pipeline steps and guarantees quicker data consumption by the analysis tools. In legacy formats, usually, the process is sequential from sequencing to variant calling, possibly including streaming over the network. Ultimately consumption of the data, which is in genomics, the analysis of the data, is delayed with regards to a scenario, like the one made possible by MPEG-G, where each step can start without waiting for the previous ones to be completely done. For example, when the alignment is completed, even on a tiny fraction of DNA, the corresponding aligned data can be sent through the network, and geneticists can immediately start their analysis.
Why is it important for the laboratory?
As already mentioned in our previous article on the compression benefit, the sizes of genomic datasets are increasing. From Sanger Sequencing, developed in the 1970s, allowing the analysis of few DNA fragments at a time (length < 1000bp) to Next-Generation Sequencing (NGS)-based methods, which have progressively allowed the parallel sequencing of multiple genes – in so-called gene panels – to whole exomes and ultimately for entire genomes. Today the growth of data puts laboratories under pressure in terms of processing time and is directly connected with its costs for this data.[5][6]
So for the following use case of MPEG-G’s Selective Access, we would like to showcase the time benefit from MPEG-G for analyzing multiple regions for the CFTR gene amongst a Whole-Exome Sequencing (WES) dataset compared to the legacy format BAM. The comparison is for the total processing time – sorting, indexing, and accessing the data. The BAM format needed 426,1 seconds – 7 minutes and 6 seconds – however, the MPEG-G file was processed in 1.6 seconds, which amounts to 99.6% of less time spent using the MPEG-G format.[7][8]
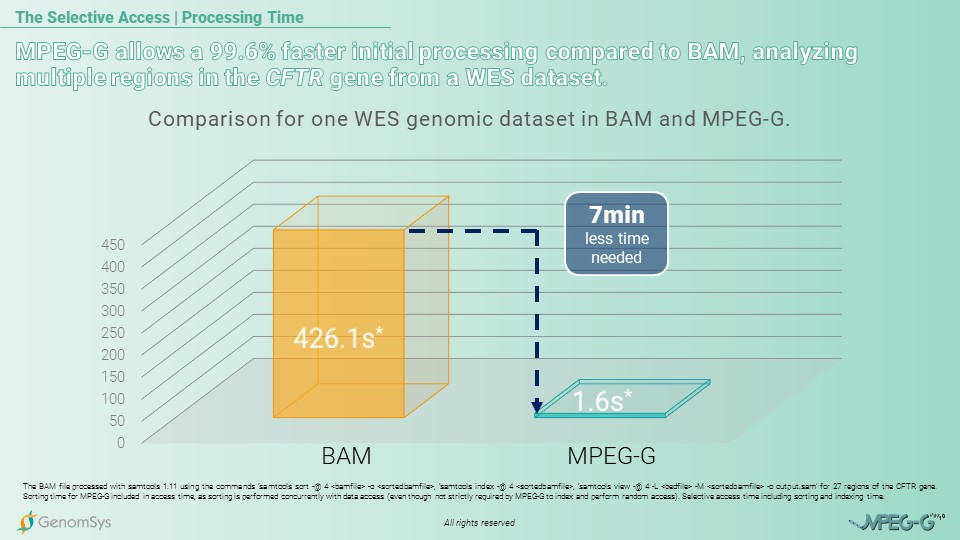
Graphic 2 – Comparison of BAM and MPEG-G for total processing time – sorting, indexing, and accessing – for multiple regions in the CFTR gene from a WES dataset.[7][8]
The benefit even increases when using a Whole-Genome Sequencing (WGS) dataset. Analyzing the same multiple regions of the CFTR gene from a WGS dataset took the BAM-formatted file 3’630 seconds – 1 hour and 30 seconds – whereas a MPEG-G formatted WGS dataset was processed in 1.1 seconds, allowing a 99.9% faster processing time.[7][8]
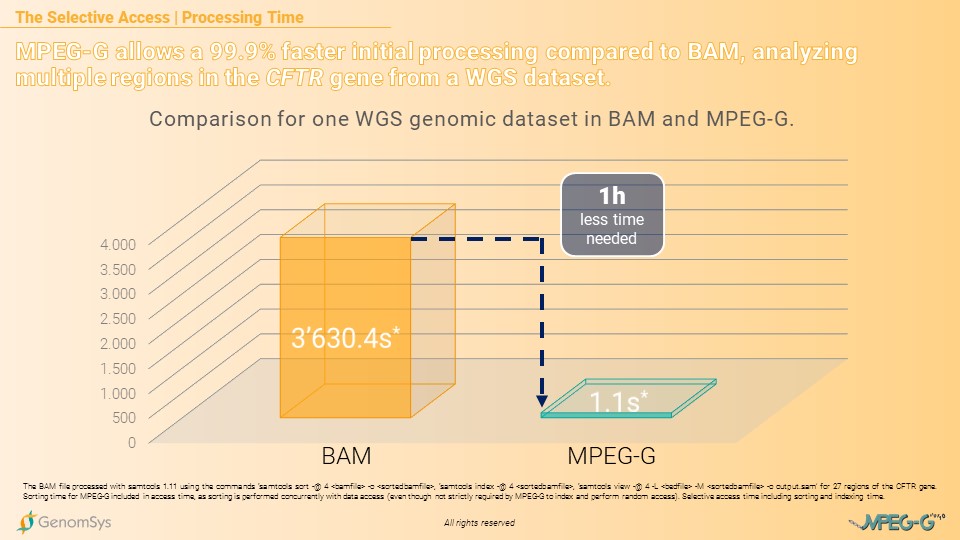
Graphic 3 – Comparison of BAM and MPEG-G for total processing time – sorting, indexing, and accessing – for multiple regions in the CFTR gene from a WGS dataset.[7][8]
GenomSys professional’s solution leveraging the compression benefit from MPEG-G
The CE marked GenomSys Variant Analyzer is natively running on the MPEG-G standard and allows seamlessly converting FASTQ data, coming directly from the sequencing machine, into MPEG-G before each analysis. This feature enables genomic laboratories to leverage the Selective Access benefit from the unique ISO standard firsthand and save precious time before starting the genomic analysis.

Graphic 4 – GenomSys professional solution workflow for genomic laboratories in their analysis of genomic data.
Also, working natively on MPEG-G, the entire analysis pipeline, including steps working on aligned data, can take advantage of all its features, from compression to Selective Access, thus minimizing the overall processing time and the storage and memory footprints.
Please find our Fact-Card on MPEG-G with information on the Selective Access Benefit and a specific use-case in the following.

Stay tuned for our next article in the series “MPEG-G, can I eat it?” on the benefits of the MPEG-G’s native feature of interoperability next week.
By Lucas Laner and Daniele Renzi on March 15th, 2022.
[1] Nina Bai and Dana Smith; The Mysterious 98%: Scientists Look to Shine Light on Our Dark Genome (2017). https://www.ucsf.edu/news/2017/02/405686/mysterious-98-scientists-look-shine-light-our-dark-genome
[2] Jamuar SS, Tan EC. Clinical application of next-generation sequencing for Mendelian diseases. Hum Genomics 2015 Jun 16;9:10-015-0031-5.
[3] Saelaert, M., Mertes, H., De Baere, E. et al. Incidental or secondary findings: an integrative and patient-inclusive approach to the current debate. Eur J Hum Genet 26, 1424–1431 (2018). https://doi.org/10.1038/s41431-018-0200-9
[4] National Computer Conference and Exposition (1957)
[5] Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 1975 May 25;94(3):441-448.
[6] Jamuar SS, Tan EC. Clinical application of next-generation sequencing for Mendelian diseases. Hum Genomics 2015 Jun 16;9:10-015-0031-5.
[7] The BAM file processed with samtools 1.11 using the commands ‘samtools sort -@ 4 <bamfile> -o <sortedbamfile>, ‘samtools index -@ 4 <sortedbamfile>, ‘samtools view -@ 4 -L <bedfile> -M <sortedbamfile> -o output.sam’ for 27 regions of the CFTR gene.
[8] Sorting time for MPEG-G included in access time, as sorting is performed concurrently with data access (even though not strictly required by MPEG-G to index and perform random access). Selective access time including sorting and indexing time.
Picture Source: xresch/ pixabay

