
Over the past decade, there has been a significant surge in data generation worldwide, with the global digital data volume growing by more than 900% since 2013, and this trend is expected to continue exponentially.[1] Technological advancements in this century, particularly in the genomics field, have enabled us to shed more light on our DNA as a basis for discovering and understanding ourselves. Population genomics researchers can now compare thousands of genomes within a population to gain a better understanding of the human body and the evolution of our DNA. However, dealing with the massive amount of data poses its own set of challenges. Therefore, it is necessary to equip scientists who work towards scientific progress with the right tools to continually improve our health.
In the following sections, we will delve deeper into population genomics, the impact of next-generation sequencing technology, and how a standardized approach that supports the handling of large genomic datasets can accelerate scientific progress in decoding our genetic code.
What is Population Genomics?
Population genomics involves using genomic technologies on a large scale to study populations of individuals, focusing on heritable traits across different spatial and temporal dimensions to gain a deeper understanding of evolutionary change.[2] This field examines the causes and mechanisms behind the changes in allele and genotype frequencies within and between populations over time, providing insight into the relationships between ethnic groups and how ancestral backgrounds are shaped by environmental factors.[3] By identifying combinations of DNA variations that may cause disease in one ethnic group but not affect another, population genomics can enhance the significance of genetic testing and improve genetic variation databases, especially when considering ethnic origin.
Population genomics has, ever since Darwin, been of interest. However, scientists such as Lewontin and Krakauer (1973) were restricted in the early days due to the sequencing capabilities. They compared multi-locus datasets from multiple populations and identified non-neutral or outlier loci by contrasting patterns of population divergence among genetic regions. All this was done with cumbersome methods that covered only small areas of DNA. But the evolution of genomic testing, due to the transformation to NGS methodologies and greater computational power, allow now to study patterns of genomic divergence on an unprecedented scale.[4]
Today, multiple initiatives around the globe, like the 100’000 genomes project in the United Kingdom, the Turkish Genome Project and Dubai Genomics, aim to collect genomes from their population to gain the beforementioned insights to understand their population’s DNA better. The overall aim is to improve the genomic databases of variations to push forward personalized medicine, through genomics, for better healthcare.
The impact of NGS – delivering the basis of a broader comparison
Since NGS, next-generation sequencing, was introduced in 2005, it allows for parallel sequencing of multiple genes.[5] A quantum leap for genomics to translate biological information into digital for more than just some small areas on the DNA.
Genomics has been transformed by the switch from Sanger sequencing, which only allowed the analysis of a few DNA fragments at a time (length < 1000 base pairs), to next-generation sequencing, which enables parallel sequencing of multiple genes and generates significantly larger genomic datasets. While Sanger sequencing was previously the gold standard and could produce only a few Gigabytes within a couple of months, sequencing today generates a data flow of multiple Terabytes each week and can soon do so within days, given the ability to sequence genomes faster. The size of a whole-genome sequencing (WGS) dataset is determined by various factors, such as the format and specific attributes (e.g., genetic area coverage, sequencing kit, sequencing machine, and number of samples within the sequencing run).
The ever-increasing adoption of NGS-based methods in clinical settings has dramatically advanced the identification of the genetic causes of Mendelian phenotypes today, providing support for diagnostic, preventive, and therapeutic strategies, and supporting the development of personalized medicinal approaches is expected in upcoming years.[6][7] But with the advent of routinely sequencing whole genomes, researchers in population genomics will now be able to compare DNA data on a broader scale.
The foundation is built to receive the entire sequence of DNA. In 2002, almost the entire genome was decoded, missing “only” 8%. But in March this year, scientists achieved the complete decoding of the human genome just perfectly in time for the 20th anniversary of uncovering the sequence of the DNA.[8] Now, it seems to be only a matter of time before enough genomes will be sequenced and compared to each other to discover further information needed during genomic testing. But as in any other industry handling many data, Big Data itself produces challenges. The use of NGS brings concerns and drawbacks associated with the demanding process of storing, processing, analyzing, and interpreting of the massive amount of generated genomics data.4
And although the evolution of sequencing has been very rapid, another bottleneck has emerged that is not in the wet lab but challenges the IT framework. Precious time is spent processing the datasets, ultimately cutting the researchers’ time in their actual work of interpreting genomic data. To compensate for precisely this unnecessary lost time, the Moving Picture Experts Group (MPEG) started back in 2016 developing the first and only open standard specification, published by ISO, for the more efficient compression and transport of next-generation sequencing data.[9]
Particularly for Population Genomics, the MPEG-G standard can deliver benefits in terms of faster transmission, less storage need and faster access to the desired region amongst the DNA sequence or the annotated results.
The compression benefit of MPEG-G
When talking about multiple whole-genome sequencing datasets, the size for such information is, to say the least high. Depending on its coverage depth and compression, a single human WGS can account for about 60 – 70 GB in legacy formats. Now in the case of population genomics, the number of datasets is more in the range of hundreds of thousands to millions.
Here is where one of the main benefits of the ISO/IEC 23092 MPEG-G standard comes into play: the efficient compression for genomic datasets. Thus far, the widely used legacy formats have been developed mainly in a setting of a couple of whole genomes, primarily developed within their own habitat of bioinformatics on an academic level for past research purposes with fewer data and smaller data size.
The MPEG-G standard is built to compress the genomic data more efficiently to provide genomic organizations with a format that allows them to store their data in smaller files resulting in fewer costs and faster transmission in an ever more connected world. So, as the beforementioned example of the 100’000 genomes project, the storage benefit, by using MPEG-G compared to the gzipped FASTQ format for the genomic data would amount to 5’217 Terabytes which in the case of cloud storage with AWS services would amount to monthly cost savings of USD122’600.[10]
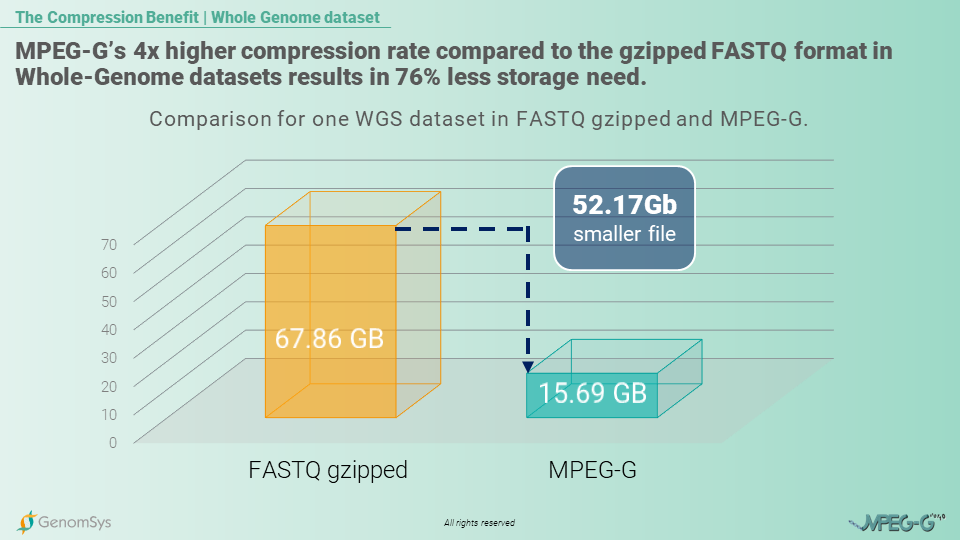
Graphic 1 – Comparison of size for a single WGS genomic datasets in gzipped FASTQ and MPEG-G.[11]
So, smaller files would benefit researchers in Population Genomics to handle the full information of a whole genome but with fewer costs for storage and, overall, faster transmission of the files. This MPEG-G benefit thus makes handling an enormous amount of data – and in population genomics, we talk about thousands of genomes – more efficient in terms of monetary and time resources.
The Selective Access benefit of MPEG-G
Another key advantage is MPEG-G’s Selective Access, with the ability to access data selectively instead of sequentially. This feature is vital for accessing specific regions within the raw DNA sequence – such as chromosomes, genes or even single bases – or scanning for variations in the annotated section. Researchers can then access the data selectively and not sequentially, which is common in legacy formats, and data processing becomes faster. Mainly the time benefit substantially grows with the size of the files.[12] But how is MPEG-G able to access data selectively and not sequentially?
The core of the Selective Access lies within the indexing structure of MPEG-G, which is natively embedded with the data. The indexing step and the sorting step – although sorting in MPEG-G is not strictly required – are inherent parts of the encoding process. Hence they can be performed concurrently with the alignment, for example, without any overhead in terms of processing time: this allows later rapid access to the desired genomic region without the need to use external indexing information to be built before accessing the data.
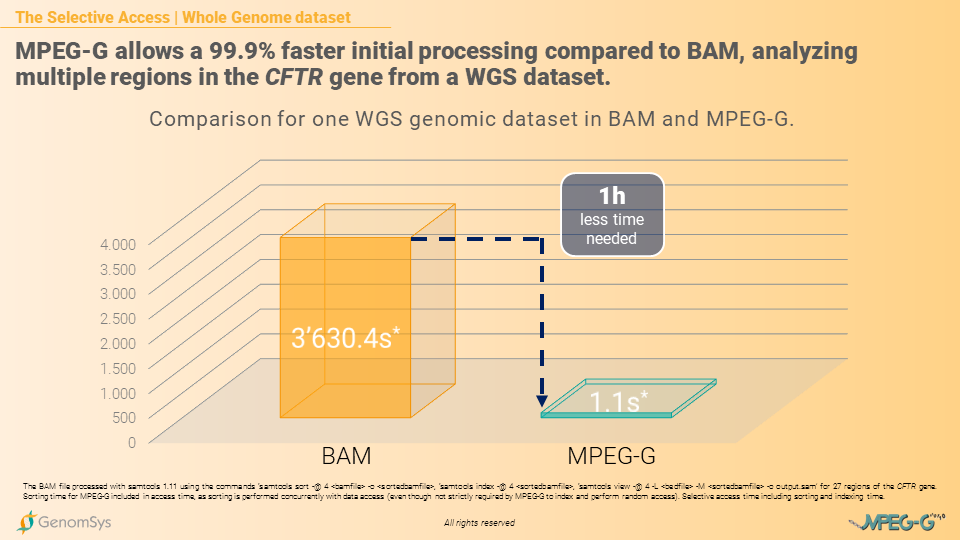
Graphic 2 – Comparison of processing time for WGS genomic datasets in BAM and MPEG-G.[13]
Also, MPEG-G datasets can be transmitted with minimum delay between data generation and consumption thanks to its access unit structure. So each access unit is an independent data chunk and can be as small as needed, making the consumer able to process data as soon as the first access unit is received, thus minimizing the overall data processing time in the genomic pipeline.
This “transport feature”, which is similarly used in streaming videos today by companies like Netflix, avoids being stuck with long waiting times between different pipeline steps and guarantees quicker data consumption by the analysis tools. In legacy formats, usually, the process is sequential from sequencing to variant calling, possibly including streaming over the network. Ultimately consumption of the data, which is in genomics, the analysis of the data, is delayed with regards to a scenario, like the one made possible by MPEG-G, where each step can start without waiting for the previous ones to be completely done. For example, even on a tiny fraction of DNA, the corresponding aligned data can be sent through the network, and researchers in population genomics can immediately start comparing datasets to find similarities amongst vast amounts of data.
GenomSys professional’s solution leveraging the interoperability feature from MPEG-G
Our solutions are designed to operate natively in the MPEG-G format, making them an ideal choice for genomics labs seeking to leverage this innovative and efficient genomic standard.
GenomSys developed a unique multi-infrastructure genomic platform compliant with ISO/IEC 23092 (MPEG-G, “the MP3 for genomic data”), the only ISO open standard for genomic data, with CE-marked as In-Vitro Diagnostic Medical Device components to support genomic data handling organizations in the processing of these data. Our platform reduces overall costs (storage needs -70%), speeds data transmission and analyses, facilitates data reuse and accelerates time to market for advanced applications.
GenomSys MPEG-G Toolkit is part of this platform. It consists of CE-marked software tools to support organizations in implementing the MPEG-G standard and leveraging its features (e.g., smaller files, faster transmission,…). Furthermore, it includes state-of-the-art bioinformatic pipelines – MPEG-G native as well – providing efficient secondary analysis of genomic data, easy to integrate into sequencing machines and/or tertiary analyses applications.
GenomSys helps organizations handle large amounts of genomic data to store them cost-efficiently and be equipped for the new era of genomics with an ever-increasing volume of data. Via GenomSys MPEG-G Toolkit, a collection of CE-marked software tools to process genomic data compliant MPEG-G, allows for encoding losslessly genomic datasets into the new genomic standard, and organizations can leverage the benefits directly.
Furthermore, the Toolkit comes with an intuitive interface to encode and decode the information as well can be implemented by GenomSys in an automatic process directly connected with the sequencing machine, even further pushing the acceleration of processing time.
References:
[1] Michael Kroker; Big Data sorgt schon 2016 für Speicher-Engpass; 2020 fehlt Speicher-Volumen von 6 Zetabytes (2015). https://blog.wiwo.de/look-at-it/2015/05/05/big-data-sorgt-schon-2016-fur-speicher-engpass-2020-fehlt-speicher-volumen-von-6-zetabytes/
[2] A. Amorim, Population Genetics, Editor(s): Stanley Maloy, Kelly Hughes, Brenner’s Encyclopedia of Genetics (Second Edition), Academic Press, 2013, Pages 407-411, ISBN 9780080961569, https://doi.org/10.1016/B978-0-12-374984-0.01195-5
[3] Charles Rotimi; POPULATION GENOMICS (2022). https://www.genome.gov/genetics-glossary/Population-Genomics
[4] Nosil, P. & Buerkle, A. (2010) Population Genomics. Nature Education Knowledge 3(10):8
[5] Jamuar SS, Tan EC. Clinical application of next-generation sequencing for Mendelian diseases. Hum Genomics 2015 Jun 16;9:10-015-0031-5.
[6] Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet 2014 Jan;59(1):5-15.
[7] Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet 2015 Aug 6;97(2):199-215.
[8] Evan Bush; A human genome has finally been fully decoded (2022). https://www.nbcnews.com/science/science-news/human-genome-finally-fully-decoded-rcna22029
[9] Moving Picture Experts Group – Genomics; MPEG-G ISO/IEC 23092 (2022). https://mpeg-g.org/
[10] In-House calculation of the same 100’000 WGS dataset with 30x coverage in each format: FASTQ gzipped 67.86GB; MPEG-G file=15.69G; AWS S3 standard pricing=0.0235USD per GB and month
[11] In-House measurements of the same WGS dataset with 30x coverage in each format: FASTQ gzipped file pairs= 67.86GB; MPEG-G file=15.69G.
[12] National Computer Conference and Exposition (1957)
[13] The BAM file processed with samtools 1.11 using the commands ‘samtools sort -@ 4 <bamfile> -o <sortedbamfile>, ‘samtools index -@ 4 <sortedbamfile>, ‘samtools view -@ 4 -L <bedfile> -M <sortedbamfile> -o output.sam’ for multiple regions of the CFTR gene. Sorting time for MPEG-G included in access time, as sorting is performed concurrently with data access (even though not strictly required by MPEG-G to index and perform random access). Selective access time including sorting and indexing time.

