
In the evolution of genomic testing, the current next-generation sequencing (NGS) methodologies have substantially increased the sequencing yield and have lowered the cost per base [1]. This development allows a faster turnaround time in genetic diagnostics, but what are the steps from the initial sampling to receiving the genomic report?
Check out our short video on the Process of Genomic Testing:
Sample Collection
Should genomic testing prove helpful in the context of disease diagnosis, prevention, or selecting appropriate treatment, sample collection will follow. Usually, the DNA is extracted from a blood sample, but genetic material can also be obtained from a saliva sample.
Sequencing
The sample is then prepared in the laboratory and afterward, the DNA sequence is determined using suitable sequencing machines. Since 2010 the NGS technology has mostly substituted the previous traditional Sanger sequencing standard developed in the late 1970s. The conventional approach only focused on single genes to be sequenced and solely one individual’s DNA at a time[2]. Thanks to NGS technology, it became routinely available to analyze multiple individuals in parallel for a set of genes with what are called gene panels. Thanks to the continuous progress of NGS technology and the piling-up of the resulting knowledge, we can analyze the entire genome today with a high degree of automation [3].
The result of the sequencing process is a file containing the individual’s sequence of the bases of their DNA for the area of interest for the particular diagnostic question.
More information on sequencing you can find here.
Analysis
Today’s genomic analysis relies heavily on computer methods and programs used to understand and use of biological and biomedical data which is referred to as Bioinformatics. A deeper explanation on Bioinformatics you can find here.
During the analysis, a complex series of bioinformatic processes are carried out to align the sequence obtained from the individual to a human reference genome and determine variations in the DNA sequence of the individual.
The human reference genome was first published in 2003 by the Genome Reference Consortium (GRC) and has been updated continuously since then, providing genetic professionals a more consistent reference in their quest to detect variations on the DNA in the diagnostic [4].
These genetic variants can involve single or multiple bases, and in some cases, disrupt specific biological functions turning out as disease-causing [5]. After identifying genetic variants, an expert provides an interpretation availing of a comprehensive set of resources or annotation databases for genetic data. The knowledge in these databases can provide information on the role that identified variants might play in specific diseases and finally lead to a report with supporting information for further medical action.
The knowledge associated with genetic variation is constantly growing. It is a lengthy effort by the entire research community worldwide to obtain reliable data to base medical decisions regarding prevention or treatment.
Over the last decade, the efficiency along this process chain for genomic testing has increased significantly. The sequencing step has become a highly automated process, directly impacting the subsequent analysis. The processing of this massive amount of electronic data can be very time-consuming. Currently, the digital pre-processing and accessing regions on the DNA sequence in legacy formats – especially when analyzing whole-exome and whole-genome sequencing datasets – can take up to multiple hours. Resulting in a delay for the start of the analysis as well as for the interpretation and ultimately slowing down the delivery of the needed report for the patient.
The ISO/IEC 23092 standard for genomic data (MPEG-G) published by ISO is ideal for improving time efficiency along the processing chain for genome sequencing data. This open international standard is structured so that it leads to a minimal time delay until data consumption. Thereby this data format contains so-called access units that can be opened and processed simultaneously. Compared to legacy formats, which can only process data sequentially, MPEG-G shortens the processing time considerably.
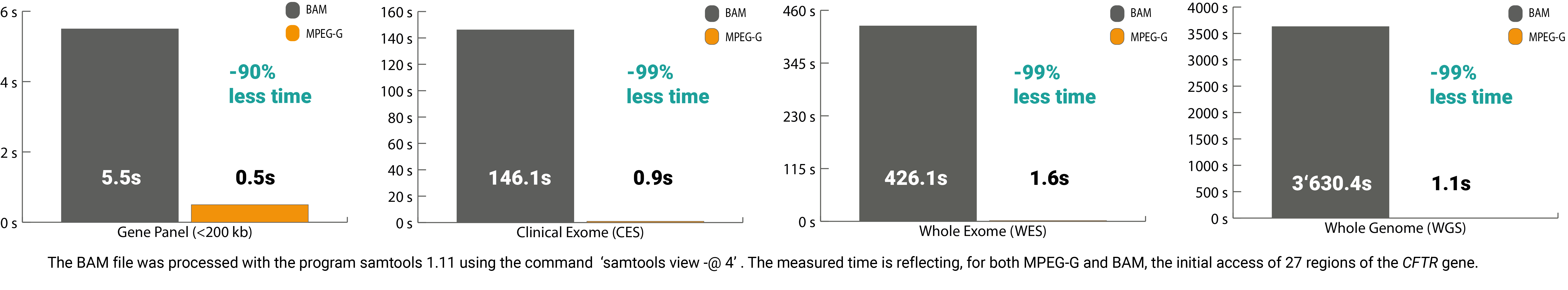
In addition, the selective access feature of the standard also delivers benefits in terms of time. The core lies in the indexing structure of MPEG-G, which allows quick access to the desired offset without spending time sorting and indexing, resulting in massive time savings. For example, the processing time to access 27 regions of interest in the CFTR gene from a whole-exome sequencing file is only 2secs vs. 450secs with legacy formats.
These benefits are now paired with the intuitive design and a wide range of filtering options, adjustable to the individual needs, included in GenomSys Variant Analyzer. Genetic experts develop this genomic analysis software for genetic experts in laboratories to deliver rapid and reliable results to help improve patients’ lives.
By Lucas Laner on December 02, 2021.
References:
[1] Buermans HPJ, den Dunnen JT. Next-generation sequencing technology: Advances and applications. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease; From genome to function 2014 10/01;1842(10):1932-1941.[2] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3917434/
[3] https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Fact-Sheet
[4] “UCSC Genome Bioinformatics: FAQ”. https://genome.ucsc.edu/FAQ/FAQreleases.html#release1
[5] Trotta L.. Genetics of Primary Immunodeficiency in Finland https://helda.helsinki.fi/bitstream/handle/10138/278894/TROTTA_e-thesis_19122018.pdf?sequence=1&isAllowed=y
Picture: mohamed Hassan/ pixabay

